





بالإضافة إلى التكنولوجيا، لطالما كان تخليق الجليكوسيدات محل اهتمام علمي، نظرًا لشيوعه في الطبيعة. وقد تناولت أوراق بحثية حديثة لشميدت وتوشيما وتاتسوتا، بالإضافة إلى العديد من المراجع المذكورة فيها، مجموعة واسعة من إمكانيات التخليق.
في تخليق الجليكوسيدات، تُدمج مكونات متعددة السكريات مع النيوكليوفيلات، مثل الكحولات أو الكربوهيدرات أو البروتينات. إذا تطلب الأمر تفاعلًا انتقائيًا مع إحدى مجموعات الهيدروكسيل في الكربوهيدرات، فيجب حماية جميع الوظائف الأخرى في الخطوة الأولى. من حيث المبدأ، يمكن للعمليات الأنزيمية أو الميكروبية، نظرًا لانتقائيتها، أن تحل محل خطوات الحماية الكيميائية المعقدة وإزالة الحماية من الجليكوسيدات بشكل انتقائي في بعض المناطق. ومع ذلك، نظرًا للتاريخ الطويل للجليكوسيدات الألكيلية، لم تُدرس الإنزيمات في تخليق الجليكوسيدات وتُطبق على نطاق واسع.
بسبب قدرة أنظمة الإنزيمات المناسبة وتكاليف الإنتاج المرتفعة، فإن التخليق الأنزيمي للبولي جليكوسيدات الألكيلية ليس جاهزًا للترقية إلى المستوى الصناعي، ويتم تفضيل الطرق الكيميائية.
في عام 1870، أبلغ ماكولي عن تخليق "أسيتوكلورهيدروز" (1، الشكل 2) عن طريق تفاعل الدكستروز (الجلوكوز) مع كلوريد الأسيتيل، مما أدى في النهاية إلى تاريخ طرق تخليق الجليكوسيد.
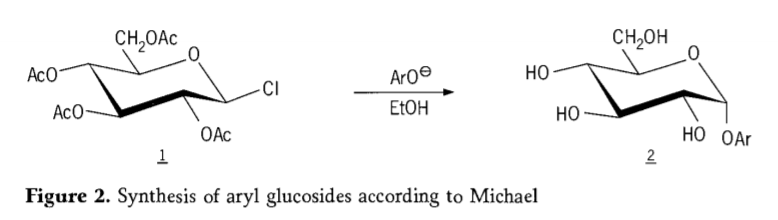
لاحقًا، وُجد أن هاليدات رباعي-أ-أسيتيل-غلوكوبيرانوسيل (أسيتوهالوجلوكوزات) تُعدّ وسيطات مفيدة في التخليق الفراغي الانتقائي لجلوكوزيدات الألكيل النقية. في عام ١٨٧٩، نجح آرثر مايكل في تحضير جليكوسيدات أريلية محددة وقابلة للتبلور من وسيطات كولي وفينولاته. (Aro-، الشكل ٢).
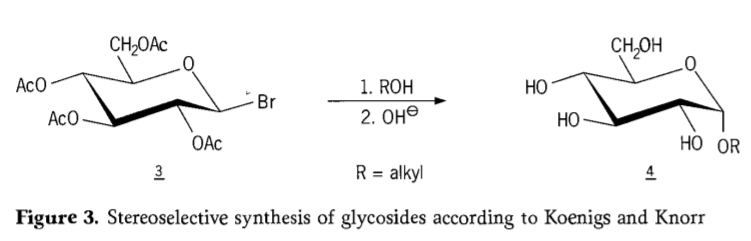
في عام ١٩٠١، بدأ مايكل في تخليق مجموعة واسعة من الكربوهيدرات والأغليكونات الهيدروكسيلية، عندما قدّم دبليو. كونيغز وإي. كنور عملية الغليكوزيدية الفراغية الانتقائية المُحسّنة (الشكل ٣). يتضمن التفاعل استبدال SN2 عند ذرة الكربون الأنيومرية، ويستمر بشكل فراغي مع عكس التكوين، مُنتجًا على سبيل المثال ألفا-غلوكوزيد ٤ من بيتا-أنومر للوسيط أسيوبروموغلوكوز ٣. يحدث تخليق كونيغز-كنور بوجود مُحفّزات الفضة أو الزئبق.
في عام ١٨٩٣، اقترح إميل فيشر نهجًا مختلفًا تمامًا لتخليق الجلوكوزيدات الألكيلية. تُعرف هذه العملية الآن باسم "غليكوزيدة فيشر"، وتتضمن تفاعلًا حمضيًا بين الجلوكوزيدات والكحولات. مع ذلك، ينبغي لأي سرد تاريخي أن يتضمن أيضًا أول محاولة مُبلغ عنها لـ أ. غوتييه عام ١٨٧٤، لتحويل الدكستروز إلى إيثانول لا مائي بوجود حمض الهيدروكلوريك. بسبب تحليل عنصري مُضلّل، اعتقد غوتييه أنه حصل على "ثنائي الجلوكوز". أثبت فيشر لاحقًا أن "ثنائي الجلوكوز" الذي ابتكره غوتييه كان في الواقع جلوكوسيد إيثيلي بشكل أساسي (الشكل ٤).
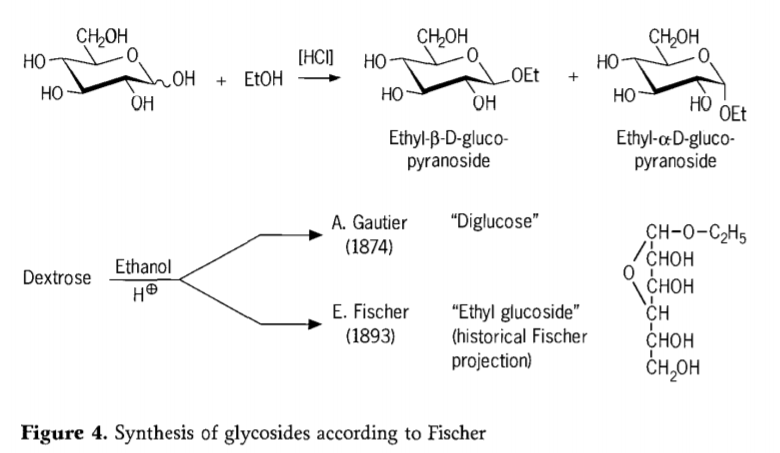
عرّف فيشر بنية إيثيل غلوكوزيد بشكل صحيح، كما يتضح من الصيغة التاريخية للفورانوزيدية المقترحة. في الواقع، نواتج الغليكوزيدية لفيشر هي في الغالب خلائط متوازنة من أنومرات ألفا/بيتا ومتزامرات بيرانوسيد/فورانوسيد، والتي تتكون أيضًا من أوليغومرات غليكوزيدية مرتبطة عشوائيًا.
وبناءً على ذلك، يصعب عزل أنواع جزيئية منفردة من مخاليط تفاعل فيشر، وهو ما مثّل مشكلةً كبيرةً في الماضي. بعد بعض التحسينات في طريقة التخليق هذه، اعتمد فيشر لاحقًا طريقة التخليق كونيغز-كنور في أبحاثه. باستخدام هذه الطريقة، كان إي. فيشر وب. هيلفيريش أول من أبلغ عن تخليق غلوكوزيد ألكيل طويل السلسلة يُظهر خصائص خافضة للتوتر السطحي عام ١٩١١.
منذ عام ١٨٩٣، لاحظ فيشر بدقة خصائص أساسية للجليكوسيدات الألكيلية، مثل ثباتها العالي تجاه الأكسدة والتحلل المائي، خاصةً في الأوساط القلوية القوية. وتُعدّ هاتان الخاصيتان قيّمتين في استخدامات البولي جليكوسيدات الألكيلية في المواد الخافضة للتوتر السطحي.
لا تزال الأبحاث المتعلقة بتفاعلات الجليكوسيدات جارية، وقد طُوّرت مؤخرًا العديد من الطرق المثيرة للاهتمام لتحضير الجليكوسيدات. يُلخّص الشكل 5 بعض إجراءات تخليق الجليكوسيدات.
بشكل عام، يمكن تقسيم عمليات الجليكوسيد الكيميائية إلى عمليات تؤدي إلى توازنات معقدة للأوليغومرات في تبادل الجليكوسيل المحفز بالأحماض.
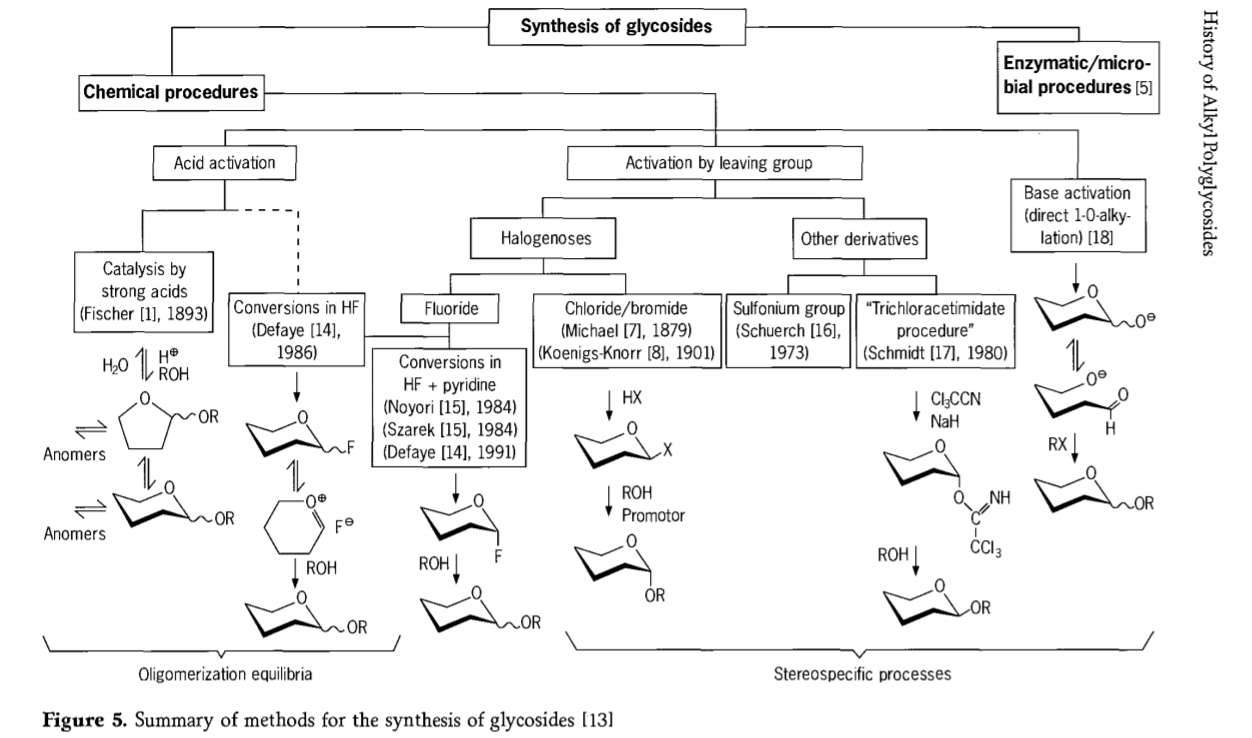
تفاعلات على ركائز كربوهيدراتية مُنشَّطة بشكل مناسب (تفاعلات فيشر الجليكوسيدية وتفاعلات فلوريد الهيدروجين (HF) مع جزيئات كربوهيدراتية غير محمية) وتفاعلات استبدال متحكم بها حركيًا، وغير رجعية، وتعتمد بشكل أساسي على التجسيم. قد يؤدي نوع ثانٍ من الإجراءات إلى تكوين أنواع فردية بدلًا من خليط معقد من التفاعلات، خاصةً عند دمجها مع تقنيات مجموعات الحفظ. قد تترك الكربوهيدرات مجموعات على ذرة الكربون المنفصلة، مثل ذرات الهالوجين، أو السلفونيل، أو مجموعات ثلاثي كلورو أسيتيميديدات، أو تُنشَّط بواسطة قواعد قبل تحويلها إلى إسترات ثلاثية الفلات.
في حالة الغليكوزيدات في فلوريد الهيدروجين أو في مخاليط فلوريد الهيدروجين والبيريدين (بولي بيريدينيوم [فلوريد الهيدروجين])، تتشكل فلوريدات الجليكوزيل في الموقع وتتحول بسلاسة إلى جليكوسيدات، كما هو الحال مع الكحولات. وقد تبين أن فلوريد الهيدروجين وسط تفاعل نشط للغاية وغير متحلل؛ ولوحظ تكثيف ذاتي متوازن (أوليغومرات) مشابه لعملية فيشر، على الرغم من أن آلية التفاعل مختلفة على الأرجح.
الجليكوسيدات الألكيلية النقية كيميائيًا مناسبة فقط لتطبيقات خاصة جدًا. على سبيل المثال، استُخدمت الجليكوسيدات الألكيلية بنجاح في الأبحاث الكيميائية الحيوية لتبلور بروتينات الأغشية، مثل التبلور ثلاثي الأبعاد للبورين والباكتيرورودوبسين بوجود أوكتيل بيتا-دي-غلوكوبيرانوسيد (أدت تجارب أخرى مبنية على هذا العمل إلى حصول دايزنهوفر وهوبر وميشيل على جائزة نوبل في الكيمياء عام ١٩٨٨).
خلال تطوير بولي جليكوسيدات الألكيل، استُخدمت طرق انتقائية فراغية على نطاق مختبري لتصنيع مجموعة متنوعة من المواد النموذجية ودراسة خصائصها الفيزيائية والكيميائية. ونظرًا لتعقيدها، وعدم استقرار المواد الوسيطة، وكمية مُهدرات العمليات وطبيعتها الحرجة، فإن عمليات التصنيع باستخدام طريقة كونيغز-كنور وغيرها من تقنيات المجموعة الوقائية قد تُسبب مشاكل تقنية واقتصادية كبيرة. أما عمليات فيشر، فهي أقل تعقيدًا نسبيًا وأسهل تنفيذًا على نطاق تجاري، وبالتالي، تُعتبر الطريقة المُفضلة لإنتاج بولي جليكوسيدات الألكيل على نطاق واسع.
وقت النشر: ١٢ سبتمبر ٢٠٢٠